firestar help page
firestar[1,2] makes predictions of functionally important residues using the large inventory of functionally important residues in the FireDB[3] database. The reliability of the transfer of functional information between the functionally important residues in FireDB and the query sequence is evaluated via the local residue conservation between the two sequences.
User Input
The user must unambiguously identify a protein sequence. Three options are allowed:- PDB code: each entry in the PDB is identified by a four letter code (e.g. "1tco"). PDB files usually contain more than one chain, so the user is prompted to give the chain identifier.
- Uploading a PDB formatted coordinates file: Here again the user is prompted to choose the chain if more than one appears in the file.
- Sequence input: At present we only accept FASTA formats. In this case some options will be disabled because no structure can be used for structural alignments.
Prediction summary
The first page returned by the server reports a compact summary of firestar analysis. An output example is shown below: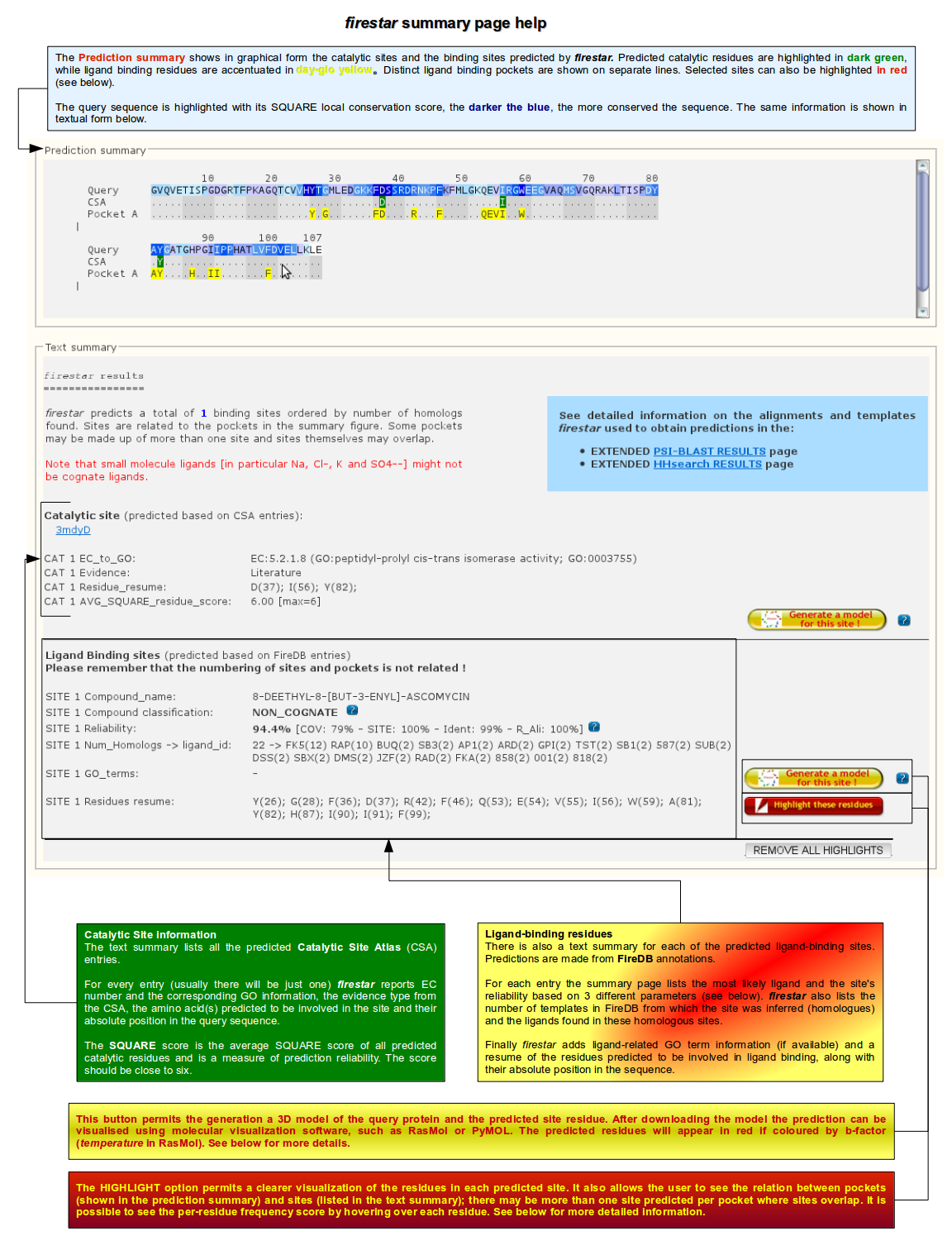
An intermediate yet informative step can be displayed following the EXTENDED links. All alignments between the query and templates found with PSI-BLAST[4] and HHsearch[5] are graphically represented hereafter, separately. Here you can find a detailed help page with all the information described: EXTENDED Help.
Biologic Relevance
In FireDB (and of course in firestar) all the small molecule binding annotations are culled from from the experimental data stored in the PDB database. PDB ligands have been divided in three categories: COGNATE, POSSIBLE_COGNATE and NON_COGNATE in order to distinguish between possible biologically important compounds and other molecules (inhibitors, analogues, solvents from the crystalization conditions, etc.)."COGNATE" compounds are those that are almost always candidates to be the real biological ligands. While "POSSIBLE COGNATE" ligands are also sometimes biological ligands, they often appear in a structure because they are present in buffers or crystallization solutions. "NON COGNATE" ligands could be drugs, inhibitors, analogues etc. etc. that are almost never natural biological ligands. Although "POSSIBLE COGNATE" and "NON COGNATE" ligands may not perform biological roles in a target protein, their binding may still provide information about the query protein, especially if the binding site is highly conserved.
If you are interested in a specific compound tag, complete lists are available here.
Per-Site Reliability
The reliability for each site is calculated combining three parameters:- COVERAGE [COV]: this is calculated with respect to the size of the binding sites annotated in FireDB; metal binding sites are expected to have very high coverage;
- Mean SQUARE' score [SITE]: The mean SQUARE score over the predicted amino acids for the site;
- IDENTITIES [Ident]: Greater weight is given to sites inferred from alignments with templates that are close homologues;
- Alignment ratio [R_Ali]: each site is predicted from at least one alignment between the target sequence and a FireDB template. The Alignment Ratio is calculated from the ratio between the number of alignments used to predict each site and the largest number of alignments that go into predicting any site for that target
3D Models
firestar selects the best template found in the HHsearch analysis that contains all the predicted site's residues and builds a model with MODELLER. B-factors in the file are modified, in order to permit user visualize predicted residues in red using visualization software, such as RasMol or PyMOL.Per-residue Frequency Score
The "Highlight Option" permits you to highlight predicted residues with their normalized relative frequency score. These scores are calculated from the alignments used to predict each site and weights depending on the evolutive similarity of the aligned template are used. The scores for the residues can be seen by moving the mouse over each individual residue. These scores are independent for each site and should not be compared between sites.High Throughput Mode
firestar is available as a REST web service. With a "wget" command you can directly query our web server and you will retrieve all the information provided in the summary page output in a easy-to-parse format. A query example is shown here:$> wget -O your_output_file_here 'http://firedb.bioinfo.cnio.es/Php/fstarTEXT.php?target=XXXXX&sequence=ZZZZZZ'
where the parameters are:
- target = an ID for your query protein;
- sequence = the entire one letter code aminoacid sequence of your protein;
- format=text; If you want to receive a HTML output, you have to set the format option as 'html'. Plain text format ouput is the default one.
e.g: $>wget -O your_ouput_file_here 'http://firedb.bioinfo.cnio.es/Php/fstarTEXT.php?target=XXXXX&sequence=ZZZZZZ&format=html' - cutoff=XXX[numeric parameter 0-100]; this option allows you to filter out all sites with a reliability score lower than the selected cut-off (default is 0).
e.g: $> wget -O your_ouput_file_here 'http://firedb.bioinfo.cnio.es/Php/fstarTEXT.php?target=XXXXX&sequence=ZZZZZZ&cutoff=50' - csa=[YES|NO|ONLY]; this option allows you to include or not predicted catalytic residues culled from CSA annotations.
You can also decide to retrieve only these and exclude all others (default is YES).
e.g: $>wget -O your_ouput_file_here 'http://firedb.bioinfo.cnio.es/Php/fstarTEXT.php?target=XXXXX&sequence=ZZZZZZ&csa=ONLY' - cog=[YES|NO]; 'NO' parameter allows you to exclude from the prediction NON_COGNATE binding sites (default is 'YES').
e.g: $>wget -O your_ouput_file_here 'http://firedb.bioinfo.cnio.es/Php/fstarTEXT.php?target=XXXXX&sequence=ZZZZZZ&cog=NO'
PLEASE, if you have a very big dataset (more than 1000 proteins), contact us and we can provide you a dedicated cluster for the analysis (dcerdan@cnio.es).
firestar is also integrated in APPRIS webservices. This web service differs from the web server in that it predicts only ligand binding residues and a confidence score for each residue.
There is a Perl program example, firestar_runner.pl that allows to use these web services to get firestar's prediccions in a simple way. It uses the following libraries:
- File::Basename
- Getopt::Long
- Data::Dumper
- JSON (Conda version)
- LWP (Conda version)
- LWP::Protocol::https (Conda version)
To run this script:
firestar_runner.pl
-i <Amino acid sequences file as FASTA format>
-o <Output file where firestar reponses will be saved>
Using this input sample, you will run the script as below:
firestar_runner.pl -i sample.faa -o sample.output